Universal Decomposition of Perturbation Effects
In this tutorial, we will systematically analyze a perturb-seq dataset by decomposing the perturbation effects on to the globally defined principal axes of cell state transitions.
[1]:
import numpy as np
import pandas as pd
import scipy
import matplotlib.pyplot as plt
import scanpy as sc
Load and preprocess the perturb-seq dataset
Load the example perturb-seq dataset. This is a CRISPRi dataset.
[2]:
adata = sc.read_h5ad('data/tutorial_data/AdamsonWeissman2016_GSM2406681_10X010.h5ad')
adata
[2]:
AnnData object with n_obs × n_vars = 65337 × 32738
obs: 'perturbation', 'read count', 'UMI count', 'tissue_type', 'cell_line', 'cancer', 'disease', 'perturbation_type', 'celltype', 'organism', 'ncounts', 'ngenes', 'percent_mito', 'percent_ribo', 'nperts'
var: 'ensembl_id', 'ncounts', 'ncells'
Normalize the total count of each cell
[3]:
sc.pp.normalize_total(adata, target_sum=1e4)
Let’s have a look at what genes are perturbed in this dataset and the number of cells for each perturbation.
[4]:
condition_col = 'perturbation'
condition_cell_counts = adata.obs[condition_col].value_counts()
for c in condition_cell_counts.index:
print(f'{c}\t{condition_cell_counts.loc[c]}')
63(mod)_pBA580 6010
Gal4-4(mod)_pBA582 1283
IER3IP1_pDS002 1222
SEC61B_pDS033 1185
ASCC3_pDS052 1142
DNAJC19_pDS026 1000
HSPA9_pDS088 894
HSPA5_pDS017 881
YIPF5_pDS186 817
SAMM50_pDS156 817
TARS_pDS405 803
XRN1_pDS411 788
GBF1_pDS043 760
TIMM23_pDS284 757
DAD1_pDS499 752
SEC61A1_pDS032 740
AMIGO3_pDS434 715
SRPRB_pDS404 703
SRP68_pDS403 699
UFM1_pDS040 692
TELO2_pDS496 691
FECH_pDS494 683
MRPL39_pDS498 683
IARS2_pDS091 673
SCYL1_pDS160 669
DARS_pDS495 664
IDH3A_pDS393 663
DERL2_pDS042 656
SEC63_pDS218 654
TTI2_pDS408 653
SYVN1_pDS442 650
SEC61G_pDS440 650
SPCS3_pDS402 647
OST4_pDS353 646
ATP5B_pDS055 644
SLMO2_pDS433 637
FARSB_pDS390 632
TMED10_pDS036 626
PSMD4_pDS488 625
EIF2B4_pDS491 625
SLC35B1_pDS046 621
EIF2B2_pDS463 615
ASCC3_pDS051 614
DDRGK1_pDS041 607
GMPPB_pDS391 606
SLC39A7_pDS219 601
SRPR_pDS482 591
TIMM44_pDS430 590
SPCS2_pDS401 587
UFL1_pDS410 585
PDIA6_pDS029 583
IER3IP1_pDS110 582
YIPF5_pDS226 574
MRGBP_pDS124 570
SCYL1_pDS159 561
P4HB_pDS397 560
MANF_pDS027 554
PTDSS1_pDS478 534
TTI1_pDS407 526
DNAJC19_pDS074 525
MARS_pDS394 523
SRP72_pDS505 520
MTHFD1_pDS395 520
EIF2S1_pDS386 511
DHDDS_pDS383 507
NEDD8_pDS396 506
TMED2_pDS175 505
GNPNAT1_pDS506 503
CARS_pDS460 501
STT3A_pDS011 499
HYOU1_pDS089 494
ARHGAP22_pDS458 490
SEC61B_pDS162 483
CHERP_pDS024 479
SARS_pDS467 473
TMEM167A_pDS038 468
KCTD16_pDS096 464
SEC61A1_pDS031 451
SOCS1_pDS479 450
AARS_pDS381 447
SEL1L_pDS373 443
IARS2_pDS090 438
EIF2B3_pDS508 415
HSD17B12_pDS087 404
DDOST_pDS382 382
CCND3_pDS006 379
QARS_pDS510 379
HARS_pDS466 325
ATF4_pBA608 325
HSPA5_pDS371 301
COPZ1_pDS462 282
CAD_pDS468 281
PPWD1_pDS398 234
COPB1_pDS065 225
GBF1_pDS044 198
EIF2AK3_pBA572 141
* 101
PSMD12_pDS008 49
ERN1_pBA575 43
ERN1_pBA574 40
XBP1_pBA579 34
PSMA1_pDS007 29
XBP1_pBA578 28
IER3IP1_pDS003 26
ATF4_pBA577 23
EIF2AK3_pBA573 20
CCND3_pDS005 18
C7orf26_pDS004 12
PSMD12_pDS009 11
ATF6_pBA586 10
YIPF5_pDS001 9
ATF4_pBA576 5
62(mod)_pBA581 2
STT3A_pDS010 1
Let’s perform a quality control. Let’s filter out perturbation with too few cells and perturbations that don’t effectively knock down the target genes.
[5]:
control_condition = '63(mod)_pBA580' # The control condition
n_cell_threshold = 100 # The minimum number of cells required for a condition to be considered
perturbation_dict = {
'condition' : [],
'perturbed_gene' : [],
'control_condition': [],
'fc': [],
'pval': [],
}
for c in condition_cell_counts.index:
# Ignore conditions with too few cells
if condition_cell_counts.loc[c] < n_cell_threshold:
continue
# Extract the perturbed genes from the condition name
perturbed_genes = c.split('_')
perturbed_genes = np.intersect1d(perturbed_genes, adata.var.index)
# Only consider single gene perturbations
if len(perturbed_genes) != 1:
continue
pg = perturbed_genes[0]
# Get the expression of the perturbed gene in the control and perturbed conditions
control_exps = adata[adata.obs[condition_col] == control_condition, pg
].X.toarray().reshape(-1)
perturbed_exps = adata[adata.obs[condition_col] == c, pg
].X.toarray().reshape(-1)
# Calculate the fold-change and p-val for the perturbed gene
fc = np.mean(perturbed_exps) / np.mean(control_exps)
pval = scipy.stats.mannwhitneyu(control_exps, perturbed_exps, alternative='two-sided')[1]
# Only keep the perturbations that are effective
if (fc < 0.8) and (pval < 1e-2):
perturbation_dict['condition'].append(c)
perturbation_dict['perturbed_gene'].append(pg)
perturbation_dict['control_condition'].append(control_condition)
perturbation_dict['fc'].append(fc)
perturbation_dict['pval'].append(pval)
perturbation_df = pd.DataFrame(perturbation_dict).set_index('condition')
perturbation_df
/tmp/ipykernel_752705/3671995774.py:33: RuntimeWarning: invalid value encountered in scalar divide
fc = np.mean(perturbed_exps) / np.mean(control_exps)
[5]:
| perturbed_gene | control_condition | fc | pval | |
|---|---|---|---|---|
| condition | ||||
| SEC61B_pDS033 | SEC61B | 63(mod)_pBA580 | 0.173211 | 0.000000e+00 |
| ASCC3_pDS052 | ASCC3 | 63(mod)_pBA580 | 0.117991 | 1.549447e-94 |
| DNAJC19_pDS026 | DNAJC19 | 63(mod)_pBA580 | 0.162940 | 0.000000e+00 |
| HSPA9_pDS088 | HSPA9 | 63(mod)_pBA580 | 0.287186 | 1.990341e-254 |
| HSPA5_pDS017 | HSPA5 | 63(mod)_pBA580 | 0.462840 | 4.543529e-96 |
| ... | ... | ... | ... | ... |
| COPZ1_pDS462 | COPZ1 | 63(mod)_pBA580 | 0.306617 | 2.228928e-69 |
| CAD_pDS468 | CAD | 63(mod)_pBA580 | 0.211891 | 2.448909e-17 |
| PPWD1_pDS398 | PPWD1 | 63(mod)_pBA580 | 0.305571 | 1.135729e-08 |
| COPB1_pDS065 | COPB1 | 63(mod)_pBA580 | 0.503718 | 1.162430e-07 |
| GBF1_pDS044 | GBF1 | 63(mod)_pBA580 | 0.331636 | 2.903447e-04 |
87 rows × 4 columns
Next, we compute the pseudo-bulk expression profile of each condition.
[6]:
from tqdm import tqdm
def get_cluster_mean_expression_matrix(adata, cluster_column):
'''Get a dataframe of mean gene expression of each cluster.'''
cluster_names = np.unique(adata.obs[cluster_column].values)
cluster_mean_df = pd.DataFrame(np.zeros((len(cluster_names), adata.shape[1]), dtype=np.float32),
index=cluster_names, columns=adata.var.index)
for c in tqdm(cluster_names):
X_c = adata[adata.obs[cluster_column] == c].X
cluster_mean_df.loc[c] = X_c.mean(axis=0)
return cluster_mean_df
# Get mean expressions of each condition
selected_conditions = np.unique(list(perturbation_df.index)
+ list(perturbation_df['control_condition']))
adata_selected = adata[adata.obs[condition_col].isin(selected_conditions)]
condition_mean_exp_df = get_cluster_mean_expression_matrix(adata_selected,
condition_col)
condition_mean_exp_df
100%|██████████| 88/88 [00:43<00:00, 2.04it/s]
[6]:
| gene_symbol | MIR1302-10 | FAM138A | OR4F5 | RP11-34P13.7 | RP11-34P13.8 | AL627309.1 | RP11-34P13.14 | RP11-34P13.9 | AP006222.2 | RP4-669L17.10 | ... | KIR3DL2-1 | AL590523.1 | CT476828.1 | PNRC2-1 | SRSF10-1 | AC145205.1 | BAGE5 | CU459201.1 | AC002321.2 | AC002321.1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 63(mod)_pBA580 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000095 | 0.003224 | 0.0 | 0.0 | 0.000555 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.000388 | 0.009653 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| AARS_pDS381 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.006999 | 0.0 | 0.0 | 0.000964 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.000000 | 0.011662 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| ARHGAP22_pDS458 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.003068 | 0.002012 | 0.0 | 0.0 | 0.000000 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.000000 | 0.011770 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| ASCC3_pDS051 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.003492 | 0.0 | 0.0 | 0.000000 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.000000 | 0.008839 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| ASCC3_pDS052 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.001639 | 0.002893 | 0.0 | 0.0 | 0.001667 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.000000 | 0.012430 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| UFL1_pDS410 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.001283 | 0.0 | 0.0 | 0.000000 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.000954 | 0.009231 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| UFM1_pDS040 | 0.000432 | 0.0 | 0.0 | 0.0 | 0.000763 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.000000 | 0.009446 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| XRN1_pDS411 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.002461 | 0.005934 | 0.0 | 0.0 | 0.000889 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.000000 | 0.011627 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| YIPF5_pDS186 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.006665 | 0.0 | 0.0 | 0.000000 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.000531 | 0.011071 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| YIPF5_pDS226 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.004654 | 0.0 | 0.0 | 0.001430 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.001212 | 0.015296 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
88 rows × 32738 columns
Now we can compute the pseudo-bulk perturbation-induced gene expression shift vector for each perturbation.
[7]:
import anndata
X_perturb = []
# Iterate through the perturbations
for c in tqdm(perturbation_df.index):
# Compute the log1p normalized mean expression of the perturbed and control conditions
control_c = perturbation_df.loc[c, 'control_condition']
x_pert = condition_mean_exp_df.loc[c].values.astype(np.float32)
x_ctl = condition_mean_exp_df.loc[control_c].values.astype(np.float32)
x_pert = np.log1p(x_pert / x_pert.sum() * 1e4)
x_ctl = np.log1p(x_ctl / x_ctl.sum() * 1e4)
# Compute the difference in mean expression between the perturbed and control conditions
X_perturb.append(x_pert - x_ctl)
X_perturb = np.array(X_perturb)
# Create the perturbation anndata
adata_perturb = anndata.AnnData(
X=X_perturb,
obs=perturbation_df.copy(),
var=adata.var.copy(),
)
adata_perturb
100%|██████████| 87/87 [00:00<00:00, 1193.11it/s]
[7]:
AnnData object with n_obs × n_vars = 87 × 32738
obs: 'perturbed_gene', 'control_condition', 'fc', 'pval'
var: 'ensembl_id', 'ncounts', 'ncells'
Project perturbation effects onto the principal axes of cell state transitions
Let’s mask out the direct target genes of each perturbation to focus on the downstream effect of the perturbed genes.
[8]:
# Mask out the direct target genes
for i in range(adata_perturb.shape[0]):
pg = adata_perturb.obs['perturbed_gene'].iloc[i]
if pg in adata_perturb.var_names:
adata_perturb.X[i, adata_perturb.var_names.get_loc(pg)] = 0
Now we can compute the gene expression program shift scores for the globally defined gene expression programs, which represent the principal axes of cell state transitions.
[9]:
# Load the pre-defined global gene expression programs
gene_exp_cluster_df = pd.read_csv('data/tutorial_data/gene_exp_program_annotation.csv', index_col=0).set_index('gene_name')
adata_perturb.var['gene_exp_cluster'] = gene_exp_cluster_df['cluster_name']
# Initialize the gene program matrix
adata_perturb.uns['gene_program_names'] = list(gene_exp_cluster_df['cluster_name'].unique())
adata_perturb.obsm['gene_program'] = np.zeros((adata_perturb.shape[0], len(adata_perturb.uns['gene_program_names'])))
# Compute the gene program matrix
for i in range(len(adata_perturb.uns['gene_program_names'])):
genes = list(gene_exp_cluster_df[gene_exp_cluster_df['cluster_name'] == adata_perturb.uns['gene_program_names'][i]].index)
genes = np.intersect1d(genes, adata_perturb.var_names)
if len(genes) == 0:
continue
scale_factor = np.sqrt(len(genes))
adata_perturb.obsm['gene_program'][:, i] = scale_factor * np.mean(adata_perturb[:, genes].X, axis=1)
Let’s define the order of the gene expression programs to plot.
[10]:
gene_program_order = [
'chromatin structure', 'DNA replication/repair', 'cell cycle (G1/S)', 'cell cycle (prometaphase)', 'cell cycle (M phase)',
'spliceosome', 'proliferation',
'ribosome biogenesis', 'ribosomal protein genes', 'mitochondrial encoded', 'cholesterol biosynthesis', 'mitochondrial ribosome',
'Golgi vesicle transport', 'unfolded protein response',
'integrated stress response', 'p53 signaling', 'lysosome/autophagy',
'pluripotency', 'Hox genes', 'glia', 'neural development', 'neuronal', 'peripheral neurons',
'visual perception', 'retinal epithelium', 'melanin biosynthesis',
'epithelial', 'respiratory epithelium', 'kidney', 'intestine', 'pancreatic', 'pancreatic islet', 'liver', 'epidermal',
'mesothelial', 'adipocyte', 'mesenchymal', 'smooth muscle', 'endothelial', 'bone',
'interferon signaling', 'TNF signaling', 'immune system', 'myeloid', 'macrophage', 'B cell', 'T cell', 'natural killer',
'mast cell', 'erythroid', 'megakaryocyte',
'muscle', 'heart', 'cilia',
]
Now we can comprehensively plot how each perturbation from this dataset push the cell state to change along each principal axis.
[11]:
fig, ax = plt.subplots(figsize=(25, 4))
# Plot a column for each expression program
for i in range(len(gene_program_order)):
x = i - 0.25 + (np.arange(adata_perturb.shape[0]) / adata_perturb.shape[0] / 2)
idx = adata_perturb.uns['gene_program_names'].index(gene_program_order[i])
y = adata_perturb.obsm['gene_program'][:, idx]
ax.axvline(i, color='lightgray', lw=0.5, zorder=-1)
ax.scatter(x, y, s=1, edgecolor='none', c='gray', rasterized=True)
# Highlight the top and bottom 3 perturbations
top_shift_indices = list(np.argsort(y)[:3]) + list(np.argsort(y)[-3:])
for j in top_shift_indices:
if np.abs(y[j]) > 0.5:
ax.text(x[j], y[j], adata_perturb.obs['perturbed_gene'].iloc[j], fontsize=10)
ax.scatter(x[j], y[j], s=6, edgecolor='red', c='none', linewidth=0.8)
#print(gene_program_order[i], adata_perturb.obs['perturbed_gene_name'].iloc[j], f'{y[j]:.2f}')
ax.axhline(0, color='lightgray', lw=0.5, zorder=-1)
ax.set_xticks(np.arange(len(gene_program_order)), gene_program_order, rotation=45, ha='right')
ax.set_xlim(-1, len(gene_program_order))
ax.set_xlabel('Gene Expression Programs')
ax.set_ylabel('Program Activity')
plt.show()
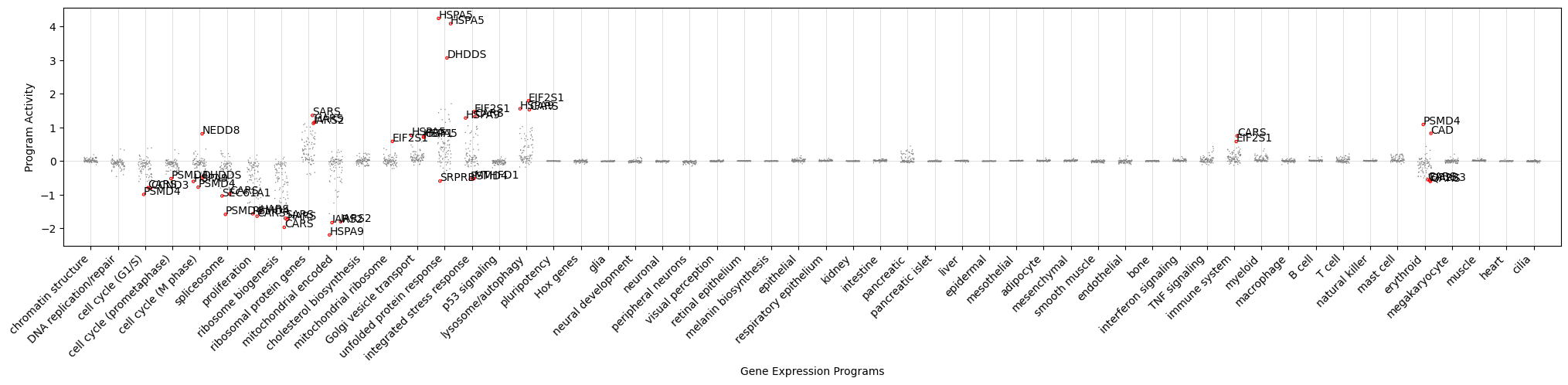
Here we can see that the major effect of perturbations are turning on stress response pathways and turning down essential programs such as spliceosome and ribosome biogenesis. But we can also find a few examples, such as PSMD4, that affect the differentiation state of the cell along the erythroid axis.
We can zoom-into the interesting genes to comprehensively visualize their perturbation effects along major axes of cell state transition.
[12]:
genes_to_show = [
'CARS', 'SARS', 'HSPA9', 'DHDDS', 'EIF2S1', 'PSMD4', 'CAD',
]
adata_to_show = adata_perturb[adata_perturb.obs['perturbed_gene'].isin(genes_to_show)].copy()
genes_to_show = [g for g in genes_to_show if g in adata_to_show.obs['perturbed_gene'].values]
gene_program_show_df = pd.DataFrame(
index=adata_to_show.obs['perturbed_gene'],
columns=adata_to_show.uns['gene_program_names'],
data=adata_to_show.obsm['gene_program']
)
fig, ax = plt.subplots(figsize=(25, 3))
import seaborn as sns
sns.heatmap(gene_program_show_df.loc[genes_to_show, gene_program_order], cmap='seismic', ax=ax, center=0,
cbar_kws={'label': 'RMS'})
ax.set_xticklabels(ax.get_xticklabels(), rotation=45, ha='right')
ax.set_xlabel('Perturbed Genes')
ax.set_ylabel('Gene Expression Programs')
[12]:
Text(283.22222222222223, 0.5, 'Gene Expression Programs')
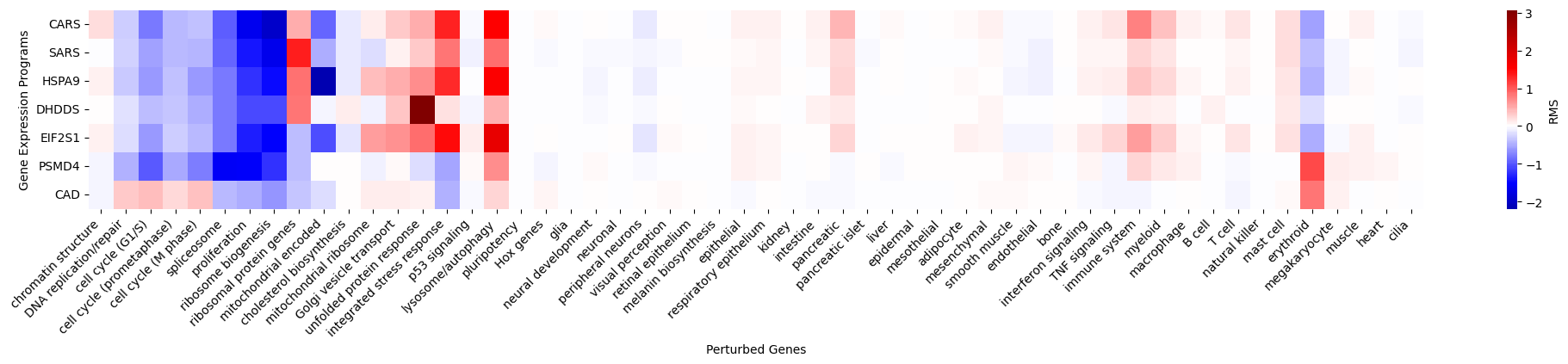
We can check the perturbation-induced expression of individual genes to validate the collective phenotypes such as the erythroid differentiation caused by PSMD4 and CAD perturbation.
Let’s plot all the genes belonging to the erythroid program.
[13]:
perturbed_genes_to_show = [
'PSMD4', 'CAD',
]
perturbed_genes_to_show = [g for g in perturbed_genes_to_show if g in adata_perturb.obs['perturbed_gene'].values]
perturbed_gene_ids = [list(adata_perturb.obs['perturbed_gene']).index(g) for g in perturbed_genes_to_show]
target_genes_to_show = sorted(adata_perturb.var[adata_perturb.var['gene_exp_cluster'] == 'erythroid'
].index.values)
target_gene_ids = [list(adata_perturb.var.index).index(g) for g in target_genes_to_show]
mtx = adata_perturb.X[perturbed_gene_ids, :][:, target_gene_ids]
fig, ax = plt.subplots(figsize=(15, 2))
sns.heatmap(mtx, cmap='coolwarm', center=0, vmin=-0.5, vmax=0.5,
xticklabels=target_genes_to_show, yticklabels=perturbed_genes_to_show,
cbar_kws={'label': 'gene expression shift'},)
plt.xlabel('Readout genes')
plt.ylabel('Perturbed genes')
[13]:
Text(158.22222222222223, 0.5, 'Perturbed genes')
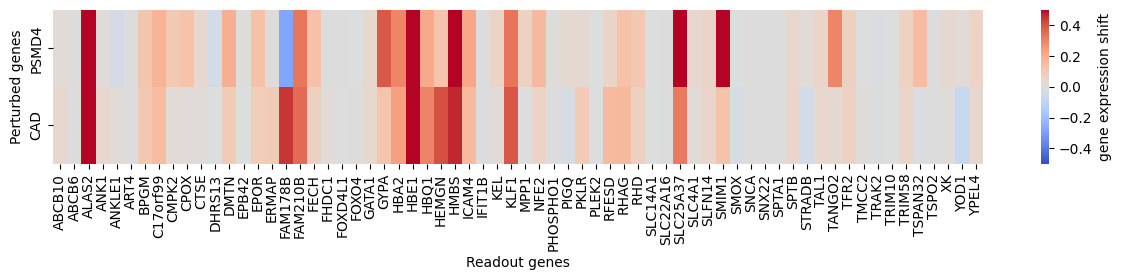
Indeed, we can see specific marker genes of erythrocytes, such as the hemoglobins (HBA2, HBE1 and HBQ1) and the transcription factor KLF1, being upregulated by knocking down PSMD4 and CAD.
Grouping of perturbations into universal clusters
So far we have analyzed the principal phenotypes of individual perturbations. We can further gain a comprehensive picture of the coupling between different cellular processes and phenotypes by grouping perturbations into the pre-defined universal clusters.
Let’s load the pre-defined clusters and compute the mean perturbation effects of each cluster.
[14]:
pert_cluster_df = pd.read_csv('data/tutorial_data/perturbation_cluster_annotation.csv', index_col=0)
pg_to_cluster_map = dict(zip(pert_cluster_df['perturbed_gene_name'], pert_cluster_df['cluster_name']))
adata_perturb.obs['cluster_name'] = adata_perturb.obs['perturbed_gene'].map(pg_to_cluster_map)
perturb_group_df = pd.DataFrame(
index=list(adata_perturb.obs[~adata_perturb.obs['cluster_name'].isna()]['cluster_name'].unique()),
columns=adata_perturb.uns['gene_program_names'],
dtype=float,
)
for pg_cluster in perturb_group_df.index:
pg_genes = list(adata_perturb.obs[adata_perturb.obs['cluster_name'] == pg_cluster].index)
perturb_group_df.loc[pg_cluster] = adata_perturb[pg_genes].obsm['gene_program'].mean(axis=0)
Let’s plot how each perturbation cluster affect the expression of the essential gene expression programs.
[15]:
pg_cluster_order = [
'chromatin organization', 'DNA replication', 'histone acetylation', 'CCT complex',
'mediator complex', 'transcription initiation', 'integrator complex', 'pol II transcription', 'pol II enlongation',
'spliceosome', 'RNA metabolism', 'RNA degradation', 'RNA methylation', 'mRNA Polyadenylation', 'mRNA surveillance',
'ribosome small unit biogenesis',
'ribosome small unit protein', 'rRNA processing 1', 'rRNA processing 2',
'ribosome large unit biogenesis', 'ribosome large unit protein',
'translation initiation', 'Recycling Of eIF2:GDP',
'protein neddylation', 'proteasome',
'protein processing in ER', 'membrane fission',
'mitochondrion organization', 'mitochondrial translation', 'mitochondrial transcription',
]
pg_cluster_order = [c for c in pg_cluster_order if c in perturb_group_df.index]
gene_program_order = [
'chromatin structure', 'DNA replication/repair', 'cell cycle (G1/S)', 'cell cycle (prometaphase)', 'cell cycle (M phase)',
'spliceosome', 'proliferation',
'ribosome biogenesis', 'ribosomal protein genes', 'mitochondrial encoded', 'cholesterol biosynthesis', 'mitochondrial ribosome',
'Golgi vesicle transport', 'unfolded protein response',
'integrated stress response', 'p53 signaling', 'lysosome/autophagy',
]
fig, ax = plt.subplots(figsize=(8, 8))
sns.heatmap(perturb_group_df.loc[pg_cluster_order, gene_program_order],
cmap='seismic', center=0, cbar_kws={'label': 'mean gene program shift'},
#xticklabels=False, yticklabels=False,
vmax=2, vmin=-2,
ax=ax
)
ax.set_xticklabels(ax.get_xticklabels(), rotation=45, ha='right')
ax.set_xlabel('Gene expression programs')
ax.set_ylabel('Functional clusters')
[15]:
Text(70.7222222222222, 0.5, 'Functional clusters')
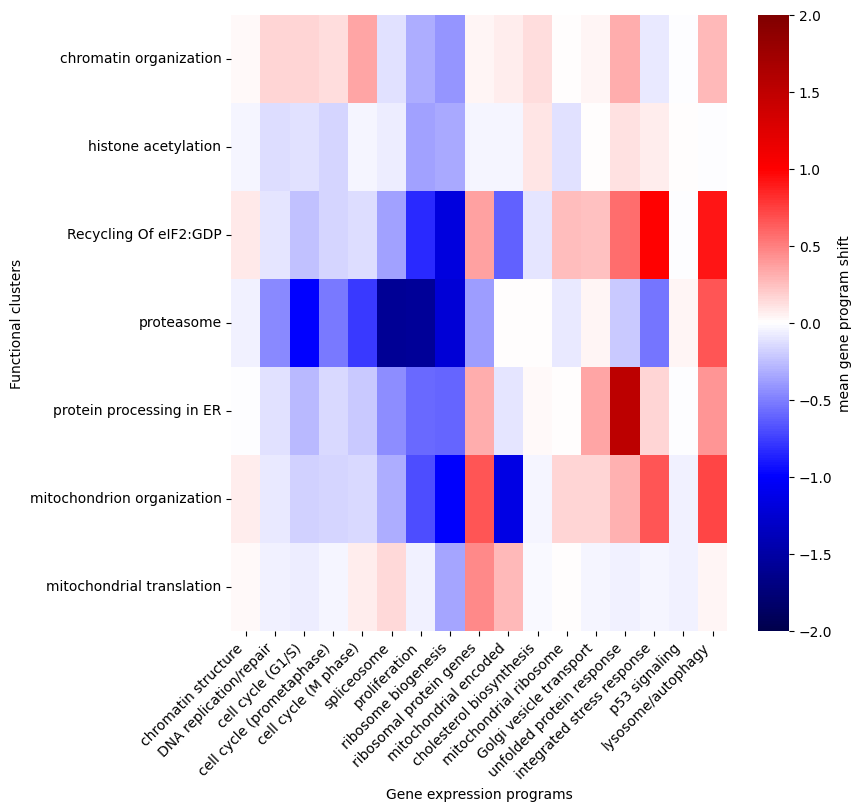
Several interesting patterns emerge:
Proliferation related processes including DNA replication/repair, cell cycle (G1/S), cell cycle (prometaphase), cell cycle (M phase), spliceosome, proliferation and ribosome biogenesis are broadly turned down by diverse perturbations, suggesting perturbation-induced cell cycle arrest.
Unlike ribosome biogenesis, which is co-regulated with genes for proliferation, the ribosomal protein genes are regulated with a different pattern, suggesting its responsibility for maintaining other aspects of cellular homeostasis.
Stress response pathways are activated by distinct spectra of perturbations. As expected, K562 cells have no p53 signaling. Several perturbation clusters, including recycling of eIF2:GDP, protein processing in ER and mitochondrion organization can activate multiple pathways, including unfolded protein response, integrated stress response and lysosome/autophagy pathways. But perturbation of proteasome selectively activate lysosome/autophagy.